Gromacs Citation
Naturally, some of those releases will be made after the year 2018 ends, but we keep 2018 in the name so users understand how up to. 2020 gromacs user survey is now live.the survey will help the gromacs developers to prioritise future gromacs developments.

Comparison of NAMD and GROMACS properties. Download Table
You can find more documentation and other material at our homepagewww.gromacs.org.

Gromacs citation. In the latter, only highly conservative fixes will be made, and only to address issues that affect scientific correctness. Recently, this method has also been applied as a scoring function in computational drug design. Here a new tool g_mmpbsa.
A package for molecular simulation and trajectory analysis,” j. It is worked on continuously, which in some. The gromacs (groningen machine for chemical simulations) software package is a program for performing molecular dynamics simulations (henceforth referred to as md simulations) [34, 35,36,37].
Nijenborgh 4, 9747 ag groningen, the netherlands. We would like input from researchers who perform any and all forms of molecular dynamics and whose experience using gromacs ranges from zero experience to expert active users. It is primarily designed for biochemical molecules like proteins, lipids and nucleic acids that have a lot of complicated bonded interactions, but since gromacs is.
Charmm27 is a widespread and popular force field for biomolecular simulation, and several recent algorithms such as implicit solvent models have been developed specifically for it. Gromacs was first developed in herman berendsens group, department of biophysical chemistry of groningen university. 5.1.2 in the mixture of 20%/80%.
(hmr) using the gromacs software. A parallel computer for molecular dynamics simulations (1993) by h bekker, h j c berendsen, e j dijkstra, s achterop, r van drunen, d van der spoel, a sijbers, h keegstra, b reitsma, m k r renardus. It is maintained by a group of developers from the universities of groningen, uppsala, and stockholm, and the max planck institute for polymer research.
It is a team effort, with contributions from several current and former developers all over world. The gromacs development teams at the royal institute of technology and uppsala university, sweden. Our use case are atomistic simulations carried out with the gromacs molecular dynamics (md) toolkit with a focus on alchemical.
Citation information when citing this document in any scientific publication please refer to it as: Gromacs is one of the most popular molecular dynamic (md) applications and is widely used in the field of chemical and bimolecular system study. We have here implemented the charmm force field and all necessary extended functional forms in the gromacs molecular si.
Advanced search include citations tables: This includes the 5.1, 2016, 2018, and 2019 release series. Gromacs is a versatile package to perform molecular dynamics, i.e.
First step in installing gromacs is to get cmake, in the terminal, type: Simulate the newtonian equations of motion for systems with hundreds to millions of particles. Gromacs usa workshop and conference 2013.
Van der spoel, “gromacs 3.0: This alert has been successfully added and will be sent to: If asked “after this operation, 16.5 mb of additional disk space will be used.
Preface and disclaimer citation information Fast, flexible and free (2005) by d van der spoel, e lindahl, b hess, g groenhof venue: Two versions of gromacs are under active maintenance, the 2019 series and the 2018 series.
Gromacs is in the public domain and distributed (with source code and documentation) under the gnu general public license. Gromacs is a versatile package to perform molecular dynamics, i.e. While this approach requires cpu resources, it has the advantage of supporting domain decomposition and all functionalities, since any special algorithm can be executed on the cpu.
I tried to observe the dynamical behavior and a protein at the surface of the mixture solvent of ethanol and water. An introduction to free energy calculations: An introduction to replica exchange simulations:
Simulate the newtonian equations of motion for systems with hundreds to. Advanced search include citations authors: The simulation was performed using gromacs ver.
Advanced search include citations tables: Computing potentials of mean force: If you want to help with developing gromacs, your are most welcome to read up on the developer guide and continue right away with coding for gromacs.
Mark a e and berendsen h j c: While we are undergoing our transition to new web pages, the releases of the source code can be downloaded here. Free energy calculation capabilities in gromacs:
Citation information when citing this document in any scientific publication please refer to it as: A global supercomputer to speed up alchemical drug design.
![]()
Running a QM/MMMD Simulation using CPMD and GROMACS
Download Gromacs For Windows 10


(PDF) The accelerated weight histogram method, AWH PMF

(PDF) Large biomolecular simulation on HPC platforms II
How to Benchmark GROMACS GPU Acceleration on HPC Clusters

How does one extend the protein simulation in Gromacs and

Performance, costs, and costefficiency for GROMACS

Download Gromacs For Windows 10

Resources HPC Software Engineering "Do as

4 reasons to publish software articles even if you’re
GROMACS—the road ahead van der Spoel 2011 WIREs
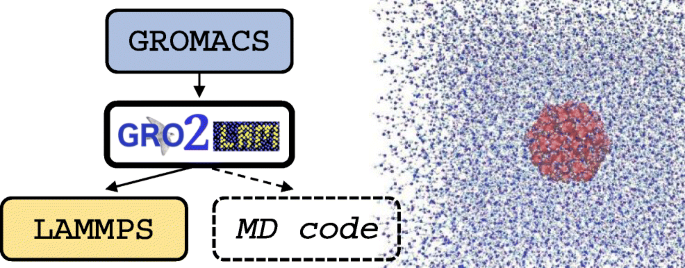
Download Gromacs For Windows 10

(PDF) GROMACS in the cloud A global to

How can we obtain an available force field parameter in

Gromacs Manual Pdf
(A) Relative performance of interpolating potentials in a

Colourcoded performance visualisation of Gromacs

A tool for simple, automated and relatively fast framework

How can I set up temperature gradient between protein and
Post a Comment for "Gromacs Citation"